API获取数据
http://rest.kegg.jp/
其中operation可选info、list、find、get、conv、link、ddi,后面的参数可以是数据库名称以及选项,在其主页有详细的介绍。
pathway map
直接使用wegt指令在服务器操作:
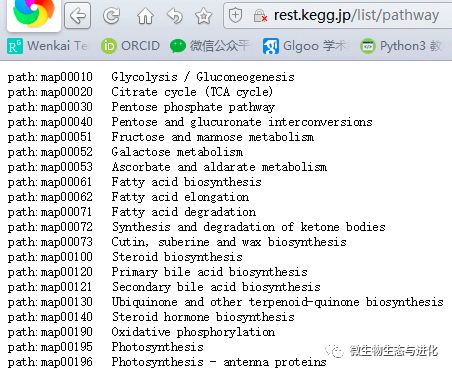
1 | wget -c http://rest.kegg.jp/list/pathway |
结果如下所示:
KO以及对应的gene
list操作可以列出所有的KO、pathway、genome,但是无法列出所有的genes(KEGG可能在这里故意设置了限制),但是我们可以通过其他方法link到genes,例如先list所有的KO,然后使用link操作获取每个KO对应的gene名称:
1 | wget -c http://rest.kegg.jp/list/ko |
结果如下所示:


其中,K02261是KO编号,COX2是基因名称,hsa是物种名称(人类), 4513为COX2在hsa中对应的编号(Entry),也就是说不同的物种对于同一个基因,在KEGG上对应的编号是不同的。如在KEGG网站中显示如下: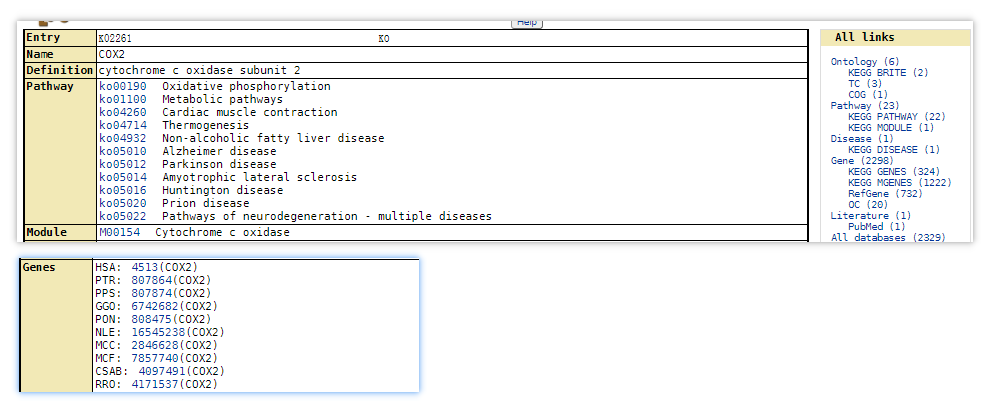
genes
如何下载所有的gene?这时候需要以基因组或者物种为单位,我们首先list所有的KEGG genome,获取所有的物种信息,然后list每个物种的gene id,如下所示:
1 | wget -c http://rest.kegg.jp/list/genome |
fasta
在下载的genome列表中还给出了每个物种的taxid,可以根据该taxid筛选特定类群的物种,这样下载更加快速。例如全部的KEGG gene有2800万个,而原核生物的大概只有1300万个,接下来我们根据gene id下载序列。获取蛋白或基因序列可以通过get操作,如下所示:
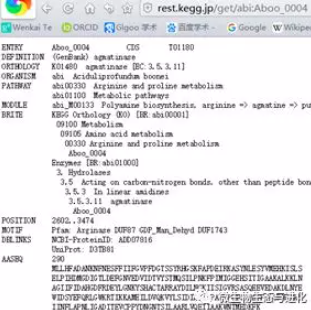
格式为genebank,里面包含基因和蛋白序列,我们可以添加选项/aaseq或者/ntseq就可以只提取序列,如下所示:

那么接下来只需写个小小的循环,遍历所有的gene id即可:
1 | cat all_genes.list | while read id; do wget -c http://rest.kegg.jp/get/$id/aaseq; done |
引用
参考文章如引起任何侵权问题,可以与我联系,谢谢。